I. García Morales
Las definiciones han ido variando a lo largo de los últimos años, sobre todo en cuanto a la duración mínima de las crisis para empezar a considerarlas como un estado epiléptico (EE). Los tiempos se han ido reduciendo y actualmente tenemos una definición más práctica en la que cualquier crisis que dure más de 5 minutos se considera que potencialmente puede evolucionar a un EE y se trata como tal, con el fin de no perder tiempo, que puede ser vital para evitar daños secundarios(1). Otras definiciones previas definían EE como crisis epilépticas de duración mayor a 30 minutos o crisis epilépticas agrupadas sin recuperación del estado neurológico previo entre las mismas durante un periodo superior a los 30 minutos.
Tipos
Independientemente de la causa, podemos distinguir dos grandes grupos en función del tipo de crisis: EE convulsivo y EE no convulsivo (Tabla I). La mayoría de las revisiones en la literatura hace referencia al tratamiento del EE convulsivo, en el que se ha alcanzado un mayor consenso en el manejo, y sobre éste se basan las recomendaciones de este capítulo.
Tabla I. Tipos de EE.
Actividad motora |
Generalizado |
Parcial |
Convulsivo |
Mioclónico Tónico Tónico-clónico |
Epilepsia parcial continua (semiología motora rítmica o pseudorítmica) |
No convulsivo |
Ausencias típicas en EGI* Ausencias atípicas en encefalopatías epilépticas Disociación electro-clínica (bajo nivel de consciencia sin actividad motora con patrón crítico en el EEG simultáneo) |
Crisis parciales complejas (más frecuente en epilepsias frontales y temporales) Crisis parciales simples (fenómenos sensitivos, autonómicos, psíquicos, etc.)
|
*EGI: Epilepsia generalizada idiopática.
EE convulsivo
Crisis con actividad tónico-clónica de más de 5 minutos de duración. El inicio puede ser focal o inicialmente generalizado. Mayor gravedad, requiere de una actuación rápida y un tratamiento agresivo para controlar las crisis, ya que pueden provocar daños irreversibles.
EE no convulsivo
Crisis sin actividad tónico-clónica de más de 5 minutos de duración. Puede tener origen focal o generalizado (ausencias): mayor controversia entre la necesidad de un tratamiento agresivo y los riesgos que puede suponer, no hay un consenso claro en cuanto al manejo.
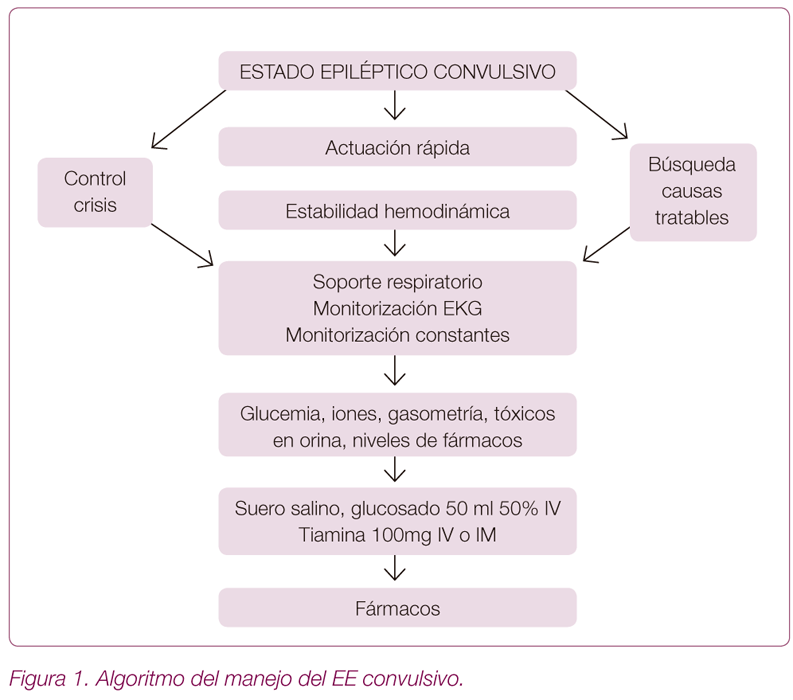
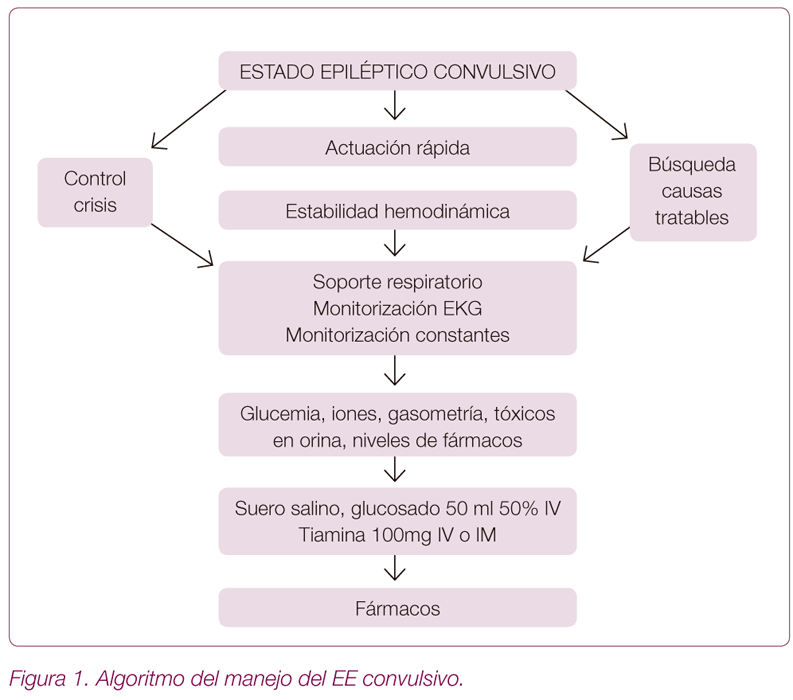
Manejo EE convulsivo (Figura 1)
Fármacos antiepilépticos
Fármacos utilizados en el tratamiento del estado de mal epiléptico(2-6) (Tabla II).
Tabla II. Fármacos para el tratamiento del estado de mal epiléptico.
1ª línea |
2ª línea |
3ª línea |
Benzodiazepinas |
Fenitoína Fosfenitoína Fenobarbital: neonatos Valproico Levetiracetam Topiramato* Lacosamida** |
Fenobarbital Fármacos nuevos Anestésicos Otros fármacos Vitaminas Cofactores |
*Topiramato tiene un mecanismo de acción en el que no se incluye el efecto sobre el GABA, cuyos receptores están bloqueados en fases finales del EE, por lo que se considera buena opción en esta fase basándose en series cortas publicadas en la literatura(2,3).
**FAE aprobado recientemente tanto en presentación oral como intravenosa, uso basado en series cortas de pacientes.
De 1ª línea
Se utilizan en la primera fase del EE (consenso Europeo)(2) (Tablas III y IV).
Tabla III. Fármacos de 1ª línea.
|
Vía de administración |
Dosis adulto |
Dosis pediátrica |
Diazepam |
Bolo iv (máximo 2-5 mg/min) Rectal |
10-20 mg 10-30 mg |
0,25-0,5 mg/Kg 0,5-0,75 mg/Kg |
Clonazepam |
Bolo iv (máximo 2 mg/min) |
1-2 mg (2 mg/min) |
200-500 µg |
Loracepam |
Vía iv (no disponible en España) |
0,007 mg/Kg, habitualmente 4 mg |
0,1 mg/Kg |
Midazolam |
Bucal, intranasal o intramuscular |
5-10 mg |
0,15-0,3 mg/Kg |
Tabla IV. Comparación entre benzodiazepinas.
|
Loracepam |
Diazepam |
Clonazepam |
Midazolam
|
Ventajas |
Acción rápida iv Acción prolongada Amplia experiencia niños y adultos Eficacia demostrada en ensayos clínicos
|
Acción rápida vía iv Vía rectal Mucha experiencia en niños y adultos
|
Acción más rápida Más potente que diazepam |
Acción rápida Solubilidad en agua Eficaz por vía oral e intranasal Bajo riesgo de acumulación |
Inconvenientes |
Tolerancia rápida Efectos adversos por solvente |
Acción corta Acumulación/ riesgo de sedación prolongada y depresión respiratoria |
Acción menos duradera que diazepam Riesgo de broncorrea |
Vida media corta |
De 2ª línea(2-6) (Tabla V)
Tabla V. Fármacos de 2ª línea.
|
Dosis inicial |
Ritmo infusión |
Dosis mantenimiento |
Fenitoína
|
Intravenosa: 10-20 mg/Kg Repetición dosis 10 mg/Kg Máximo 20-40 mg/Kg |
< 50 mg/min Habitualmente 1.000 mg en 40-45 min |
4-6 mg/Kg/día al cabo de las 12 horas de la dosis inicial |
Valproico
|
Intravenosa: 15-20 mg/Kg en 3-5 min Repetición dosis 15 mg/Kg Máximo 45 mg/Kg |
4-6 mg/Kg/min
|
0,25-1,25 mg/Kg/hora a los 30 min del bolo inicial
|
Levetiracetam
|
Intravenosa: 20mg/Kg
|
Habitualmente 500-3.000 mg en 5-15 minutos
|
20-30 mg/Kg/día al cabo de las 12 horas de la dosis inicial |
Fenobarbital (Neonatos)
|
Intramuscular:10-20 mg/Kg Intravenosa: 10-20 mg/Kg 15-30 mg/Kg en recién nacidos Repetición: 10 mg/Kg Máximo: 35-40 mg/Kg |
< 60 mg/min
|
2-4mg /Kg /día al cabo de las 12-24 horas de la dosis inicial |
Topiramato
|
600-1.600 mg /día en suspensión vía nasogástrica |
|
200-400 mg cada 12 horas al cabo de las 12-24 horas de la dosis inicial |
Lacosamida
|
Intravenoso: bolo 200 mg en 30 min (dosis variable 200-400) Mantenimiento: 200-600 al día |
|
200-400 mg cada 12 horas al cabo de las 12-24 horas de la dosis inicial |
De 3ª línea
El tratamiento se lleva a cabo en la UVI(2,3,4) (Tabla VI).
Tabla VI. Fármacos de 3ª línea.
Tiempo |
Fármaco (anestésicos) |
Medidas generales |
Monitorización |
> 60 min |
Tiopental 3-5 mg/Kg bolo; 3-5 mg/Kg/h Pentobarbital 10-15 mg/Kg; 0,1-1mg/Kg/h Midazolam: bolos de 0,2 mg/Kg (máximo 2 mg/Kg); 0,05 mg/Kg/h Propofol 1-2 mg/Kg bolo (máximo 10 mg/Kg); 2-10 mg/Kg/h |
• Cuidados intensivos: ventilación y mantenimiento hemodinámico • Control de presión intracraneal |
• ECG • EEG: control patrón brote supresión • Control iones • Control niveles de fármacos |
EE refractario
Definiciones
Se habla de EE refractario (EER) cuando las crisis continúan a pesar del tratamiento con benzodiazepinas y un antiepiléptico(4), y se considera un EE superrefractario (EESR) cuando las crisis continúan o recurren tras 24 horas o más tras el inicio del tratamiento con anestésicos, incluyendo aquellos casos que recurren ante la reducción o retirada de los fármacos anestésicos(3).
Alternativas terapéuticas
En los EE que no responden tras haber probado terapias convencionales, se pueden emplear tratamientos alternativos que tan solo han sido validados en casos o series cortas de pacientes. El uso de una terapia combinada farmacológica y no farmacológica tiene efectos aditivos y puede estar indicada en estas fases(4). Hay casos con buena recuperación tras periodos prolongados de EE, por lo que conviene seguir intentando alternativas terapéuticas (Tabla VII).
Tabla VII. Alternativas terapéuticas.
No farmacológicas |
Farmacológicas |
Cirugía |
Ketamina |
Estimulación cerebral profunda |
Lidocaína |
Estimulación magnética transcraneal |
Anestésicos inhalados |
Terapia electroconvulsiva |
|
Dieta cetogénica |
|
Pronóstico
La morbimortalidad depende de la causa subyacente y es tres veces mayor en el EER (16-39%) que en un EE no refractario(4). El riesgo de epilepsia tras un EE es tres veces mayor que tras una primera crisis y el riesgo de déficit cognitivo tiene relación con la causa. El pronóstico funcional es peor en el EER. La probabilidad de volver a la situación basal es del 21% en el EE refractario y del 60% en el no refractario(4). La edad, la etiología del EE y la duración de las convulsiones antes de ser controladas durante las dos primeras horas son factores pronóstico que determinan la evolución del paciente(8, 9).
Bibliografía
1. Meldrum BS. The revised operational definition of generalised tonic-clonic (TC) status epilepticus in adults. Epilepsia. 1999;40:123-4.
2. Shorvon S. The treatment of status epilepticus. Curr Opin Neurol. 2011;24: 165-70.
3. Shorvon S, Ferlisi M. The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain. 2011;134:2802-18.
4. Rossetti AO, Lowenstein DH. Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurol. 2011;10:922-30.
5. Stojanova V, Rossetti AO. Oral topiramate as an add-on treatment for refractory status epilepticus. Acta Neurol Scand. 2012;125(2).
6. Synowiec AS, Yandora KA, Yenugadhati V, Valeriano JP, Schramke CJ, Kelly KM. The efficacy of topiramate in adult refractory status epilepticus: experience of a tertiary care center. Epilepsy Res. 2012;98:232-7.
7. Rossetti AO, Hurwitz S, Logroscino G, Bromfi eld EB. Prognosis of status epilepticus: role of aetiology, age, and consciousness impairment at presentation. J Neurol Neurosurg Psychiatry. 2006;77:611-15.
8. Neligan A, Shorvon SD. Prognostic factors, morbidity and mortality in tonic-clonic status epilepticus: a review. Epilepsy Res. 2011;93:1-10.
9. Robert Silbergleit, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, et al. Intramuscular versus Intravenous therapy for prehospital status epilepticus. NEJM. 2012;366:591-600.